Distrofia miotónica
Sinónimos
Distrofia miotónica, enfermedad de Curschmann, enfermedad de Curschmann-Steinert
engl.: Distrofia miotónica (muscular).
Introducción
La distrofia miotónica es una de las distrofias musculares más frecuentes. Se asocia con debilidad muscular y atrofia muscular, especialmente en la región de la cara, el cuello, los antebrazos y las manos, así como en la parte inferior de las piernas y los pies. La característica aquí es la combinación de los síntomas de debilidad muscular y relajación muscular retardada después de la contracción. Además, los trastornos de otros sistemas corporales ocurren en diferentes grados: pérdida testicular y trastornos hormonales, arritmia cardíaca, opacidad del cristalino (cataratas), trastornos de la deglución y del habla, ocasionalmente el rendimiento intelectual se ve afectado en el curso de la enfermedad. La distrofia miotónica es una enfermedad hereditaria que tiende a aparecer antes de generación en generación. Se descubrió que un cambio característico en el material genético del cromosoma 19 es la causa de la enfermedad, lo que conduce a una acumulación reducida de una proteína que asegura la integridad de la membrana de la fibra muscular. En general, la gravedad de la enfermedad y la participación de varios órganos varía de un paciente a otro, y la esperanza de vida del paciente está determinada principalmente por la participación del corazón. No existe una terapia causal para la enfermedad, los síntomas relacionados con la enfermedad de los órganos afectados pueden tratarse sintomáticamente con medicamentos. Existe un mayor riesgo de anestesia durante las operaciones, que puede reducirse mediante el uso de ciertos medicamentos.
definición
La miotonía distrófica es la distrofia muscular más común en la edad adulta y se asocia con debilidad muscular, atrofia y relajación muscular retardada después de la actividad. La enfermedad también puede afectar varios otros sistemas de órganos. Clínicamente, existen 3 formas diferentes de distrofia miotónica
- un congénitoinnato) con la aparición de síntomas en la infancia
- un adulto (forma más común) a partir de la segunda y tercera décadas de la vida y
- una forma tardía que comienza a una edad avanzada.
Epidemiología
La frecuencia de distrofia miotónica se da como 1/8000 - 1/20000. La enfermedad se hereda de forma autosómica dominante, lo que significa que los hijos de los afectados tienen un riesgo del 50% de desarrollar la enfermedad ellos mismos, independientemente del sexo. Existe una tendencia a que la aparición de la enfermedad sea más temprana de generación en generación y la enfermedad sea más pronunciada ("Anticipación“).
causa principal
La causa de la distrofia miotónica es la extensión de una sección en Cromosoma 19 más allá de cierto nivel. Esto conduce a una producción reducida de una proteína que es en parte responsable de la estabilidad de la membrana de la fibra muscular. La extensión del alargamiento aumenta con la herencia de generación en generación y muestra una cierta conexión con el inicio y la gravedad de los síntomas.
Síntomas
En la forma adulta de distrofia miotónica, combinación de debilidad muscular progresiva, especialmente en los músculos de la mano y el antebrazo, el pie y la cara, con una reacción de relajación retardada de los músculos después del ejercicio (miotonía). Esto también se puede observar especialmente en los músculos de las manos y los dedos, así como en los músculos de la cara y la garganta. B. Dificultad para aflojar un puño cerrado o volver a abrir los ojos cerrados. A medida que la enfermedad progresa, puede provocar dificultad para tragar o, debido a la afectación de los músculos respiratorios, dificultad para respirar.
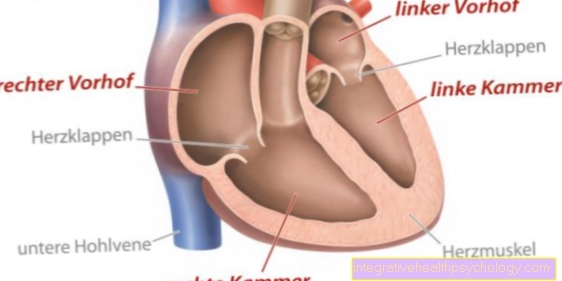
Se observan arritmias en el corazón, palpitaciones y tropiezos del corazón. En la zona de los órganos genitales aparecen síntomas como encogimiento de los testículos, menstruación ausente o irregular y complicaciones del embarazo.
Además, los pacientes a menudo sufren opacidades del cristalino (cataratas, "cataratas") y pérdida de audición del oído interno.
En la forma infantil de distrofia miotónica, los niños afectados caen temprano debido a la debilidad muscular ("bebé flexible“= Bebé recién nacido flácido), bebiendo y creciendo debilidad y retraso en el desarrollo motor. El curso de la enfermedad suele ser más severo. El curso tardío de la enfermedad puede, en determinadas circunstancias, ser atípico y, por ejemplo, solo se puede descubrir en el contexto de un diagnóstico adicional de cataratas o en el contexto de una aclaración familiar de casos de enfermedad en la descendencia directa. Los pacientes con distrofia miotónica tienen un mayor riesgo de anestesia, ya que la enfermedad puede conducir a varios anestésicos comunes con mayor frecuencia a complicaciones, especialmente en el área del sistema cardiovascular y la respiración, que en pacientes sanos. Por lo tanto, es importante que el anestesista esté informado sobre la presencia de la enfermedad antes de la operación.
Diagnósticos diferenciales
Los diagnósticos diferenciales incluyen otros trastornos miotónicos según los síntomas predominantes (relajación retardada de los músculos) u otras distrofias musculares (atrofia muscular). Además, las enfermedades del sistema nervioso pueden provocar debilidad y pérdida de los músculos controlados por los nervios afectados.
Diagnóstico
Clínicamente revolucionaria es la presencia de miotonía (relajación retardada) de los músculos con atrofia muscular (reducción de tamaño y encogimiento de los músculos). Al medir la actividad eléctrica del músculo en el EMG (electromiograma), un hallazgo característico del bajo nivel de las erupciones individuales (como en las distrofias musculares) y la aparición de series de descargas rápidas (como en la miotonía, las llamadas "Ruido del bombardero en picado).
El análisis de sangre muestra los síntomas de la muerte de las células musculares (aumento del valor de una enzima muscular, "CK") y posiblemente las consecuencias de la participación del sistema endocrino (valores disminuidos de hormonas sexuales). El examen genético humano puede detectar el alargamiento de una sección del cromosoma 19 en las células sanguíneas, que es característica de la distrofia miotónica. Si se ha realizado el diagnóstico de distrofia miotónica, es importante aclarar la función del corazón al menos mediante un EKG (electrocardiograma) para encontrar indicios de un posible trastorno funcional en una etapa temprana.
terapia
Actualmente no es posible una terapia causal para la distrofia miotónica. Las arritmias cardíacas pueden tratarse con medicación y, si es necesario, con marcapasos, y los espasmos musculares con medicación que tienen un efecto estabilizador sobre el estado de activación de la célula muscular. Si hay trastornos hormonales, estos también se pueden tratar con medicamentos. Hasta cierto punto, la fisioterapia se puede utilizar para contrarrestar los síntomas de la debilidad muscular con el fin de garantizar la movilidad del paciente y prevenir una mala postura.
pronóstico
La enfermedad progresa lentamente y varía de un paciente a otro. La esperanza de vida de los pacientes se reduce a un promedio de 50 a 60 años y a menudo se produce la muerte. Insuficiencia cardiaca a.